Concerning information relating to the identification of the Investigational Medicinal Product(s) to be used in the Clinical Trial and the IMP's status.
Once the sub-section is complete click the  button at the bottom of the page to move to the next sub-section.
button at the bottom of the page to move to the next sub-section.
Use at the foot of each screen to ensure completion of all questions in Section D.
Tip: Sections D.3.8 to D.3.10 are available via the 'add active substance' function.
NOTE: If there is no clear 'Test IMP' or '' in your study design, indicate all Investigational Medicinal Products (IMPs) as 'Test IMP'.Each strength and pharmaceutical form should be recorded as a separate Investigational Medicinal Product (use "copy IMP" and edit the strength of each active substance and/or pharmaceutical form of the IMP).
- Choose the Investigational Medicinal Product Category from the drop-down list. Click the drop-down list to select relevant option (D.1.2 and D.1.3).
Note: In Section D.2, the information provided should be that which specifically relates to the actual product being used in the trial in the Member State to which the is addressed. For example, if the CTA is submitted in France, the has a in France and Germany, and the sponsor chooses to use the registered in Germany for the purposes of the trial, it is the German trade name// number that should be mentioned in this section.
- When you answer D.2.1, if you select the 'No' option then go to D.3.
If 'Yes' then complete section D.2.1.1.1 to D.2.1.1.4.
UNLESS the IMP has a in the Member State concerned by this application, but the trade name and marketing authorisation holder are not fixed in the protocol. In such a case, complete D.2.1.2 with the name of the Member State to which the application is submitted, answer 'Yes' to D.2.1.2.1, and then go to section D.2.2.
Tip: If the Investigational Medicinal Product has a Marketing Authorisation in the Member State concerned by this application but the trade name and marketing authorisation holder are not fixed in the protocol, go to section D.2.2, below.
- If the IMP has a Marketing Authorisation in the country from which it is sourced for use in this Clinical Trial, please complete section D.2.1.1.with the information relevant to the country from which the product has been sourced.
- In free text field D.2.1.1.1, if the IMP has a Marketing Authorisation in the country from which it is sourced for use in this clinical trial, specify the Product Name as registered by the Marketing Authorisation Holder (). It is available from the , or product labelling.
- In free text field D.2.1.1.1.1, specify the EudraVigilance Product Code here when available (obtained from Eudravigilance Medicinal Product Dictionary EVMPD).
- In free text field D.2.1.1.1.2, specify the Name of the Marketing Authorisation holder, which is available from the Summary of Product Characteristics (SmPC), or Product Labelling..
Tip: For more information, see details in
EudraPharm.
- In free text field D.2.1.1.1.3, specify the Marketing Authorisation number, which is issued when a Marketing Authorisation is granted in a MS. It is available from the Summary of Product Characteristics (SmPC) or Product Labelling.
- In section D.2.1.1.4:
- Answer 'Yes' if there are any trial-specific operations that could affect the product quality, such as modification of the pharmaceutical form (e.g. over-encapsulation, colour, dilution, re-tableting for blinding etc.) or removal from the primary packaging and repacking (e.g. removal from a blister and putting in a bottle). If the blinding consists in over encapsulating tablets, trial specific coating (modified colour, or debossing), this information should be reported here.
- Answer 'No' If the product has only been relabeled or repackaged.
- In section D.2.1.2, click the drop-down list to specify the name of the country where the holder was granted the Marketing Authorisation of the actual Investigational Medicinal Product to be used in the clinical trial in the Member State concerned by the application.
- If the Investigational Medicinal Product has a Marketing Authorisation in several countries, enter the name of the country (or of one of the countries, if one of them is a Member State, choose this one) that granted the Marketing Authorisation for the actual Investigational Medicinal Product to be used in the trial in accordance with section D.2.1.1.2.
- Where the product is a Centrally Authorised Product, give the Member State in which the product was intended to be marketed (i.e. the one for which it is labelled) or, if bulk product is used, choose one of the Member States.
- In section D.2.1.2.1, answer 'Yes' if the Marketing Authorisation of the IMP to be used in the clinical trial in the Member State concerned by the application was granted by the same Member State. Answer 'No' if the Marketing Authorisation was granted by another country.
- Complete section D.2.2 when the IMP has a Marketing Authorisation in the Member State concerned by this application, but the trade name and marketing authorisation holder are not fixed in the protocol.
Tip: You should also have answered 'Yes' to D.2.1 and have completed D.2.1.2 with the name of the Member State to which the application is submitted, and 'Yes' to D.2.1.2.1.
- Section 2.2.1 should be answered 'Yes' when the protocol only identifies the INN and the investigator can use whichever brand is locally available.
The protocol of the clinical trial may specify only the INN of the product used in the trial (for example 'paracetamol') when, for the same active substance there are several different trade names available in the Member state concerned and no one of them is specified by the protocol.Note: If 'Yes', give active substance in section D.3.8 or D.3.9 from the IMP details field.
- If D.2.2.1 (above) is answered 'Yes' then D.2.2.2 may be 'Yes' or 'No'.
Select 'Yes' if, in the protocol, treatment regimens for the Investigational Medicinal Product allow different combinations of marketed products (only defined by their INN) used according to local practice at some or all investigator site in the concerned Member State (this case is frequently observed in oncology or HIV clinical trials). In this case each site might have a different combination compared to other sites.Note: If 'Yes', give active substance in section D.3.8 or D.3.9 from the IMP details field.
- If D.2.2.1 (above) is answered 'Yes' then D.2.2.3 must be 'No'. However, if the answer to this field is 'Yes' then D.2.2.1, D.2.2.2 and D.2.2.4 should be 'No'.
Note:If 'Yes', give the ATC group of the applicable authorised codes in the ATC code field (level 3 or the level that can be defined) in D.3.3
- If D.2.2.1 (above) is answered 'Yes' then D.2.2.4 must be answered 'No'.
- In D.2.3.2, if 'Yes is selected, please provide justification for using simplified dossier in the covering letter.
- In section D.2.4, the term 'authorised' should be understood in the context of Directive 2001/20/ (that is to say an authorisation, according to Directive 2001/20/EC, has been given for a trial using this Investigational Medicinal Product).
- In section D.2.4.1,add the Member States where the use of the IMP has been previously authorised.
- Multiple selection of Options:To select one value, click the value and then click 'Copy'.
To select more than one value, hold CTRL then click the other value you wish to select, then click 'Copy'.
To delete all selected values from the right-hand field, click 'Remove All' button. Note: The above technique can be used for any options organised in the same format of two fields.
- Answer D.2.5 according to the Community register on orphan medicinal products (Regulation (EC) no. 141/2000):
http://ec.europa.eu/health/documents/community-register/html/alforphreg.htm
- If the D.2.5 was answered 'Yes', please enter the orphan drug designation number in D.2.5.1.
Note: This is in reference to the regulation (EC) No.141/2000 on Orphan Medicinal Products.
- Answer D.2.6 as appropriate. If 'Yes', please answer D.2.6.1.1 and D.2.6.1.2 depending on the source of the scientific advice. This scientific advice may be given by a National Competent Authority or by the (European Medicines Agency) or both.
The sub-section is now complete.
Now click header bar to open the next sub-section (D.3 ).
 'Add IMP' to start the first Investigational Medicinal Product (IMP) or create another one.
'Add IMP' to start the first Investigational Medicinal Product (IMP) or create another one.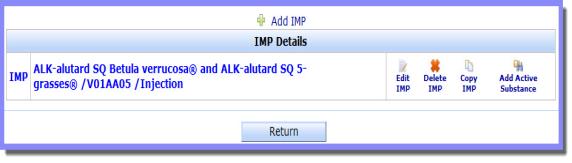 Tip: Click on the screenshot to view it full size and click on the various onscreen elements to learn more about the functionality of the screen.
Tip: Click on the screenshot to view it full size and click on the various onscreen elements to learn more about the functionality of the screen.

 on the 'D. IMP Identification' screen to find and add an active substance from the list of available active substances in the eXtended EudraVigilance Medical Product Dictionary (
on the 'D. IMP Identification' screen to find and add an active substance from the list of available active substances in the eXtended EudraVigilance Medical Product Dictionary (

